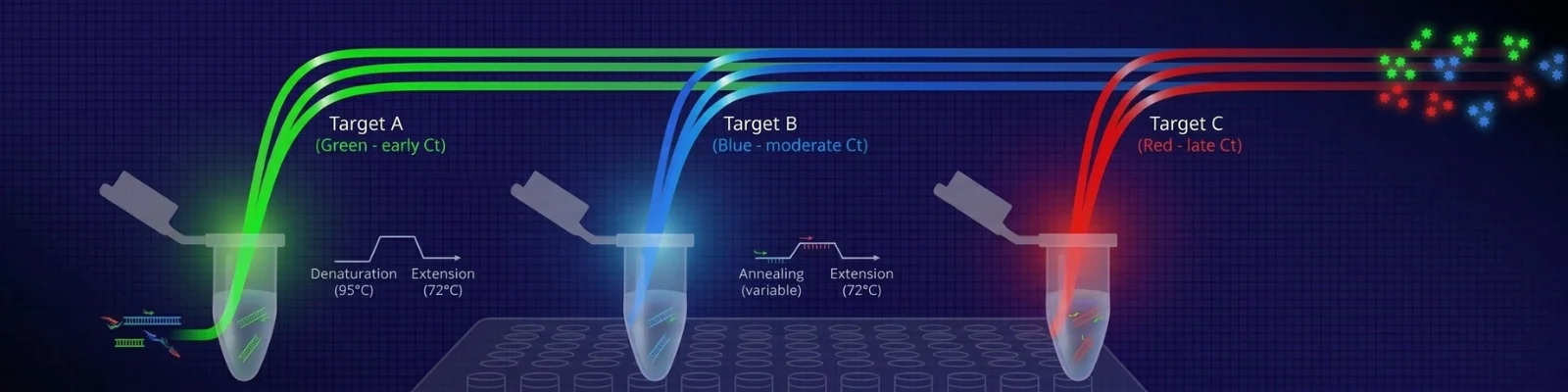
Real-time PCR (also known as quantitative PCR or qPCR) is a powerful and common technique for accurate analysis of gene expression. It is used to amplify and simultaneously quantify a targeted DNA molecule.
Unlike traditional PCR, which involves running an electrophoresis gel at the end to visualize the amplified products, real-time PCR enables the detection and quantification of nucleic acids during the PCR process.
This allows for the continuous tracking of the reaction’s progress and accurate quantification of the nucleic acids as they are being amplified.
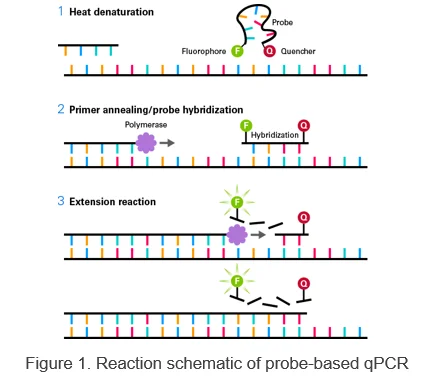
Oligonucleotides modified with a 5’ fluorophore (e.g., FAM) and a 3’ quencher (e.g., TAMRA) are added to the reaction. Under annealing conditions, the probe hybridizes in a sequence-specific manner to the template DNA.
Fluorescence of the fluorophore is suppressed by the quencher. During the extension reaction, the 5’→3’ exonuclease activity of Taq DNA polymerase degrades the hybridized probe, releasing quencher suppression and allowing fluorescence.
To maximize sensitivity, our kits use TaKaRa Ex Taq DNA Polymerase Hot-Start Version, a hot-start PCR enzyme that minimizes nonspecific amplification.
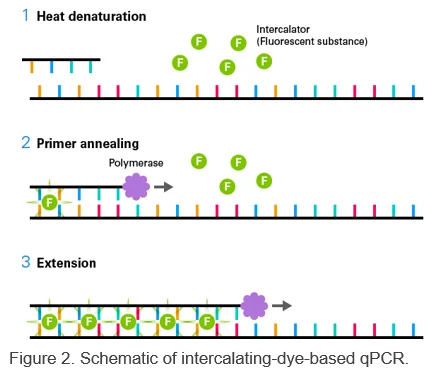
This method uses a DNA intercalator (e.g., TB Green) that emits fluorescence when bound to double-stranded DNA.
Monitoring fluorescence allows for quantification of amplification products.
Following amplification, performing a melt curve analysis provides information on the specificity of your PCR products.
To maximize specificity and sensitivity, our kits use Takara Ex Taq DNA Polymerase Hot-Start Version.
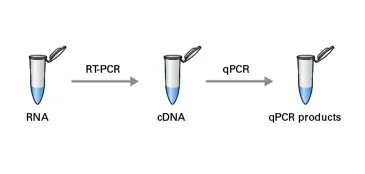
When starting with RNA samples, one must first perform a reverse transcription (RT) step to generate cDNA.
Two-step RT-qPCR performs the RT step in one tube and the qPCR reaction in a separate tube.
Uracil-DNA glycosylases (UDGs) are highly conserved enzymes involved in DNA repair across evolution.
UDG and UNG are often used interchangeably, as they both serve the same function in qPCR by preventing carryover contamination.
Preventing contamination is crucial in qPCR, as even trace amounts of DNA can lead to false positives.
To address this, UNG can degrade amplification products from prior PCR runs while leaving the original template intact.
To prevent carryover contamination in qPCR, use a master mix containing UNG or UDG.
Absolute quantification determines the absolute number (i.e., number of copies) of a target using a standard sample that has a known number of copies.
Relative quantification allows a relative comparison between samples. Typically, a target gene and a reference gene are simultaneously assayed for normalization.
Relative quantification provides the difference in expression level in the unknown sample compared with the control sample.